




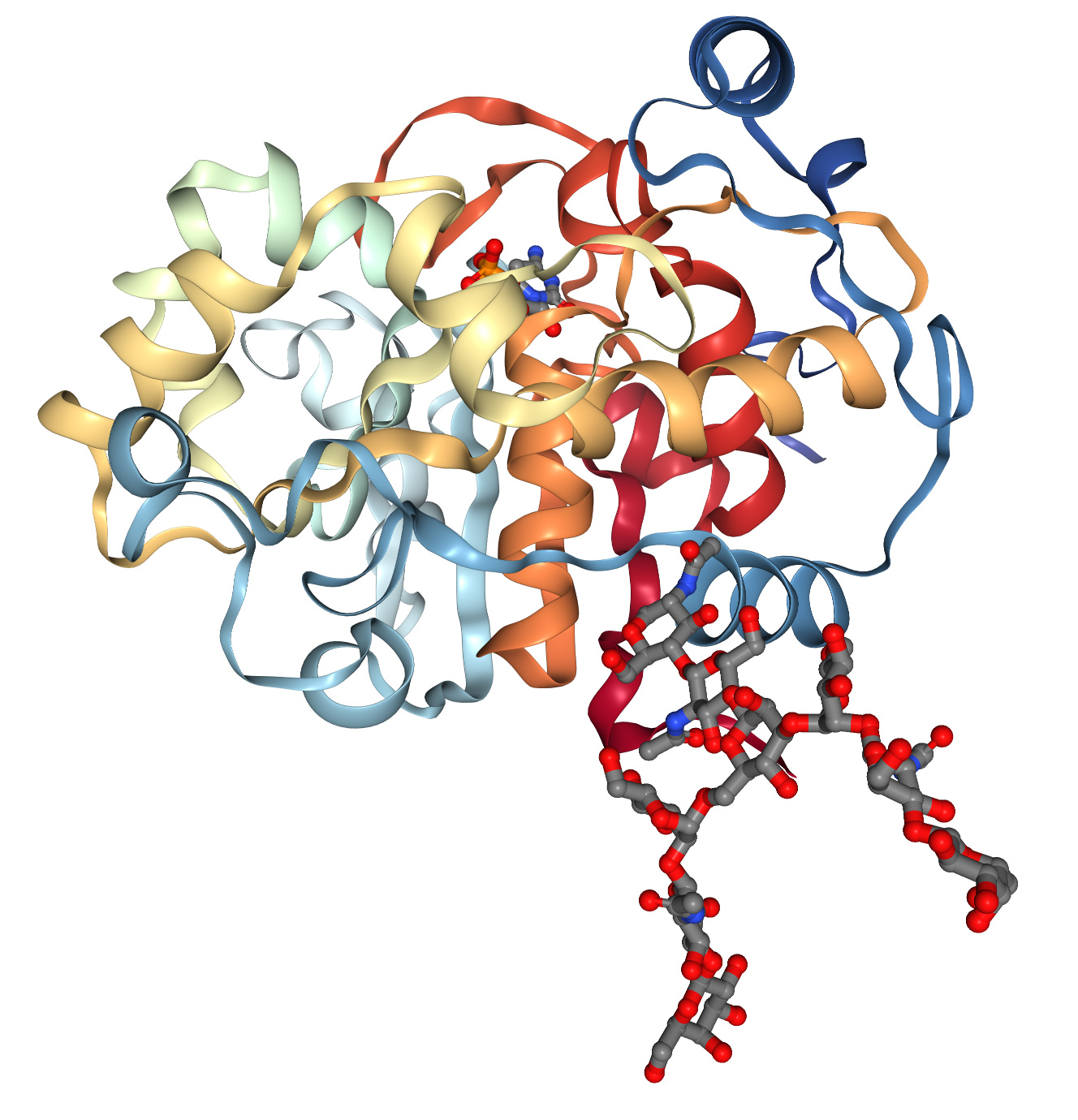
Le prix Lasker de recherche médicale fondamentale de cette année a été décerné à Demis Hassabis et John Jumper pour leurs contributions à la création du système d'intelligence artificielle AlphaFold qui prédit la structure tridimensionnelle des protéines en fonction de la séquence du premier ordre des acides aminés.
Leurs résultats résolvent un problème qui inquiète depuis longtemps la communauté scientifique et ouvrent la voie à une accélération de la recherche dans le domaine biomédical. Les protéines jouent un rôle essentiel dans le développement des maladies : dans la maladie d’Alzheimer, elles se replient et s’agglutinent ; dans le cancer, leur fonction régulatrice est perdue ; dans les troubles métaboliques congénitaux, elles sont dysfonctionnelles ; dans la mucoviscidose, elles se logent au mauvais endroit dans la cellule. Ce ne sont là que quelques-uns des nombreux mécanismes à l’origine des maladies. Des modèles détaillés de structure des protéines peuvent fournir des configurations atomiques, orienter la conception ou la sélection de molécules à haute affinité et accélérer la découverte de médicaments.
Les structures des protéines sont généralement déterminées par cristallographie aux rayons X, résonance magnétique nucléaire et cryomicroscopie électronique. Ces méthodes sont coûteuses et chronophages. De ce fait, les bases de données existantes sur les structures 3D des protéines ne comptent qu'environ 200 000 données structurales, alors que le séquençage de l'ADN a produit plus de 8 millions de séquences protéiques. Dans les années 1960, Anfinsen et al. ont découvert que la séquence 1D d'acides aminés peut se replier spontanément et de manière répétitive en une conformation tridimensionnelle fonctionnelle (figure 1A), et que des « chaperons » moléculaires peuvent accélérer et faciliter ce processus. Ces observations constituent un défi de plus de 60 ans en biologie moléculaire : prédire la structure 3D des protéines à partir de la séquence 1D d'acides aminés. Grâce au succès du Projet Génome Humain, notre capacité à obtenir des séquences 1D d'acides aminés s'est considérablement améliorée, rendant ce défi encore plus urgent.
Prédire la structure des protéines est difficile pour plusieurs raisons. Premièrement, toutes les positions tridimensionnelles possibles de chaque atome dans chaque acide aminé nécessitent une exploration approfondie. Deuxièmement, les protéines exploitent au maximum la complémentarité de leur structure chimique pour configurer efficacement les atomes. Comme les protéines possèdent généralement des centaines de « donneurs » de liaisons hydrogène (généralement l'oxygène) qui devraient être proches de l'« accepteur » de liaisons hydrogène (généralement l'azote lié à l'hydrogène), il peut être très difficile de trouver des conformations où la quasi-totalité des donneurs est proche de l'accepteur. Troisièmement, les exemples pour l'apprentissage des méthodes expérimentales sont limités ; il est donc nécessaire de comprendre les interactions tridimensionnelles potentielles entre les acides aminés à partir de séquences unidimensionnelles, en utilisant les informations sur l'évolution des protéines concernées.
La physique a d'abord été utilisée pour modéliser l'interaction des atomes à la recherche de la conformation optimale, et une méthode a été développée pour prédire la structure des protéines. Karplus, Levitt et Warshel ont reçu le prix Nobel de chimie 2013 pour leurs travaux sur la simulation informatique des protéines. Cependant, les méthodes basées sur la physique sont coûteuses en calcul et nécessitent un traitement approximatif, ce qui rend impossible la prédiction de structures tridimensionnelles précises. Une autre approche « fondée sur la connaissance » consiste à utiliser des bases de données de structures et de séquences connues pour entraîner des modèles grâce à l'intelligence artificielle et à l'apprentissage automatique (IA-ML). Hassabis et Jumper appliquent des éléments de la physique et de l'IA-ML, mais l'innovation et les progrès en termes de performance de cette approche proviennent principalement de l'IA-ML. Les deux chercheurs ont combiné de manière créative de grandes bases de données publiques avec des ressources informatiques de niveau industriel pour créer AlphaFold.
Comment savons-nous qu'ils ont « résolu » le casse-tête de la prédiction structurale ? En 1994, le concours d'évaluation critique de la prédiction structurale (CASP) a été créé. Il se réunit tous les deux ans pour suivre les progrès de la prédiction structurale. Les chercheurs partageront la séquence 1D de la protéine dont ils ont récemment résolu la structure, mais dont les résultats n'ont pas encore été publiés. Le prédicteur prédit la structure tridimensionnelle à l'aide de cette séquence 1D, et l'évaluateur juge indépendamment la qualité des résultats prédits en les comparant à la structure tridimensionnelle fournie par l'expérimentateur (fournie uniquement à l'évaluateur). Le CASP effectue de véritables évaluations en aveugle et enregistre les sauts de performance périodiques liés à l'innovation méthodologique. Lors de la 14e conférence CASP en 2020, les résultats de prédiction d'AlphaFold ont montré un tel bond en avant que les organisateurs ont annoncé que le problème de prédiction de la structure 3D avait été résolu : la précision de la plupart des prédictions était proche de celle des mesures expérimentales.
Plus largement, les travaux de Hassabis et Jumper démontrent de manière convaincante comment l'IA-ML pourrait transformer la science. Leurs recherches montrent que l'IA-ML peut construire des hypothèses scientifiques complexes à partir de sources de données multiples, que des mécanismes d'attention (similaires à ceux de ChatGPT) peuvent découvrir des dépendances et des corrélations clés dans les sources de données, et que l'IA-ML peut auto-évaluer la qualité de ses résultats. L'IA-ML est essentiellement une science.
Date de publication : 23 septembre 2023